АдеВаск: результаты исследования и перспектива регистрации

АдеВаск — единственный разработанный в России препарат, который влияет на развитие течения БАС. В настоящее время завершены клинические исследования, препарат готовится к регистрации. На Пятой пациентской конференции по БАС научный сотрудник Научного центра неврологии, к.м.н. Алексей Васильев рассказал об опыте работы с препаратом АдеВаск и результатах исследования. Публикуем расшифровку доклада спикера.
Сегодня терапевтические подходы к лечению БАС основываются на современных представлениях о гибели двигательных нейронов. Список точек приложений для разработки каких-то новых лекарственных препаратов стал достаточно обширен. Это и результаты исследований на трансгенных животных, и клинические исследования препаратов, которые используются для терапии других нейродегенеративных заболеваний, таких как болезнь Паркинсона, болезнь Альцгеймера, спинальная амиотрофия у детей, миопатии детского возраста и другие заболевания. Для терапии бокового амиотрофического склероза сегодня существует всего два одобренных препарата — эдаравон и рилузол, эффективность которых достаточно невысокая.
Методы генной инженерии
В настоящее время нейротрофические факторы мозга рассматриваются как одно из перспективных соединений, которые участвуют в регуляции и контроле за выживаемостью двигательных нейронов. К основным нейротрофическим факторам относят фактор роста эндотелия сосудов (VEGF) и ангиогенин (ANG). По данным ряда авторов было показано, что данные сосудистые нейротрофические факторы способствуют выживаемости спинальных мотонейронов в условиях ишемии, а также в пренатальном и постнатальном периоде (до и после родов — Прим. ред.).
Были проведены исследования, которые показывали, что при удалении из гена фактора роста эндотелия сосудов определенных элементов, которые определяли реакцию на гипоксию, у трансгенных мышей развивается синдром, схожий с проявлениями болезни мотонейрона у людей. Кроме того, у больных БАС были выявлены определенные корреляции уровня VEGF в спинномозговой жидкости с формой и характером прогрессирования заболевания. Например, у медленно прогрессирующих форм БАС пациенты имели более высокие концентрации уровня VEGF, чем, например, при бульбарной форме. Поэтому данные соединения были предложены в качестве возможной терапии для пациентов с болезнью мотонейронов.
Для того, чтобы доставить данные гены непосредственно в клетки-мишени или в двигательные нейроны, было предложено использовать специфические соединения — рекомбинатные аденовирусы. Они безопасны для человека, не вызывают вирусной инфекции, не встраиваются в геном клеток-мишеней и выводятся из организма в течение 3—5 недель. Также эти аденовирусы способны проникать в широкий спектр популяций двигательных нейронов и осуществлять высокую экспрессию генов в двигательных нейронах.

Существует ДНК аденовируса с областью, которая ответственна за размножение вируса. Эта область путем генно-инженерных решений удаляется, а вместо нее встраивается ген VEGF или ангиогенина. При их внутримышечном введении и максимальном захвате аксонами такие рекомбинатные вирусные частицы попадают в двигательный нейрон, где осуществляется экспрессия данных и генов и, соответственно, увеличивается уровень концентрации нейротрофических факторов.
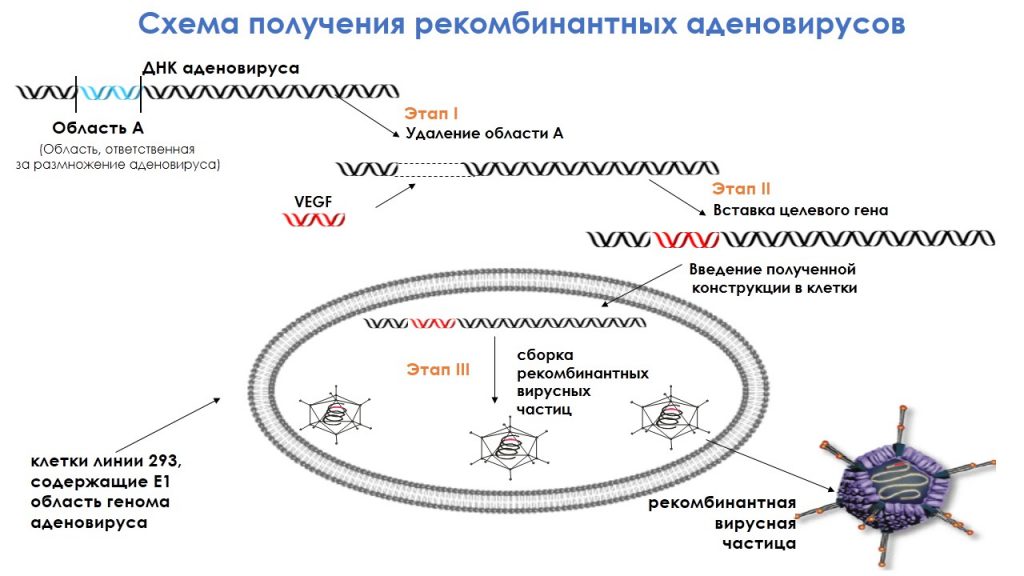
Как работает АдеВаск
Препарат АдеВаск представляет собой смесь рекомбинантных псевдоаденовирусных частиц, которые экспрессируют факторы роста эндотелия сосудов и ангиогенина человека в суммарной концентрации 7,0 на 10 в 11-ой степени частиц в 3 мл буферного раствора. Препарат вводится внутримышечно, один флакон — стандартная доза объемом 3 мл.
Целью нашего исследования было оценить безопасность и эффективность препарата АдеВаск у пациентов с боковым амиотрофическим склерозом при его многократном введении в течение 12 месяцев.
В исследование было включено 100 пациентов. Дата первого визита пациента состоялась 23 декабря 2015 года, закончилось исследование 2 марта 2018 года. Продолжительность исследования составила 53 недели, из них на скрининг было выделено 2 недели, период лечения составил 48 недель. Период последующего наблюдения, где оценивалась безопасность и развитие нежелательных явлений у пациентов, которые закончили основные 48 недель терапии, длился 3 недели. В исследовании было использовано три дозы препарата — по 1,5 мл, 3 мл и 6 мл. Все пациенты получали внутримышечные инъекции одной из доз препарата или плацебо с интервалом в три недели. Общее количество введений препарата составило 17 раз в течение 48 недель лечения.

Для оценки эффективности использовали две так называемые конечные точки. Первая — улучшилось или стабилизировалось состояние пациента по шкале ALS-FRS-R (оценка двигательных нарушений у больных с БАС). Уменьшение балла по шкале в течение одного года менее чем на 25 % или снижение балла по шкале на 12 баллов от исходного считалось достижением данной конечной точки и показателем эффективности проводимой терапии.
Вторичными переменными эффективности служили функции внешнего дыхания, жизненная емкость легких, индекс массы тела, мышечная сила в кисти, показатели ночной пульсоксиметрии, которая выявляла эпизоды ночной десатурации во время сна у пациентов. Кроме этого, оценивались выживаемость и качество жизни пациентов по европейской шкале качества жизни, а также исход в виде перевода пациентов на неинвазивную вентиляцию легких или установки гастростомы.
Для оценки безопасности проводимой терапии мы использовали совокупность методов, которые были направлены на контроль физиологических показателей, таких как артериальное давление, частота сердечных сокращений, температура, ЭКГ, которая снималась в 12 отведениях. Также контролировали показатели лабораторных исследований — клинический и биохимический анализ крови, оценивали развитие нежелательных явлений. Сравнили безопасность препарата с плацебо.
Результаты исследования
Динамика нарастания неврологического дефицита (симптомы, свойственные для местного поражения определенных структур центральной или периферической нервной системы) у больных БАС, которая оценивалась по шкале ALS-FRS-R, только в дозе 6 мл достоверно замедляла темп его прогрессирования. Остальные дозы препарата — 1,5 мл, 3 мл и плацебо — на степень нарастания дефицита влияния не оказывали.
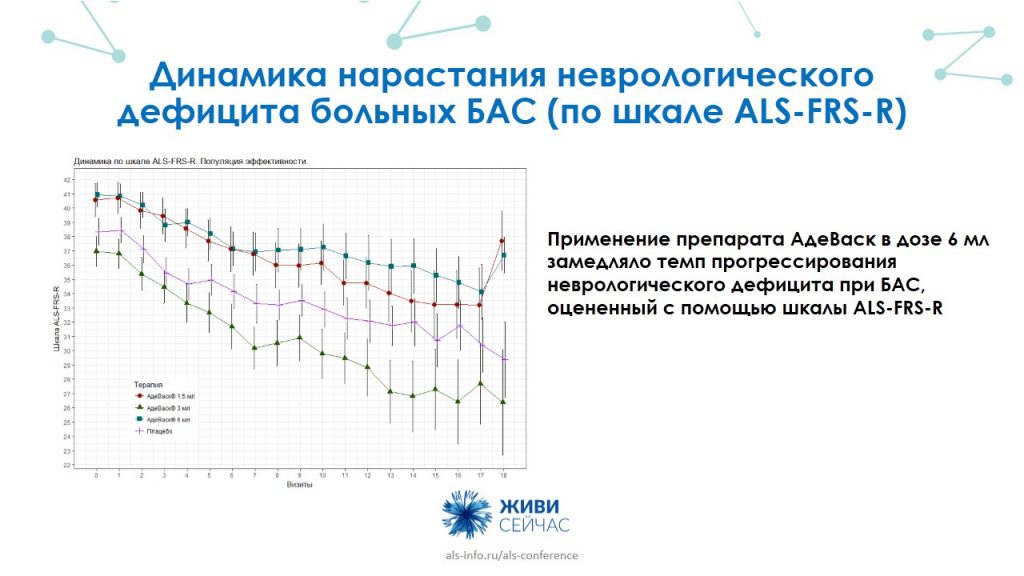
При измерении дыхательных нарушений применение препарата АдеВаск в дозе 1,5 мл и 6 мл достоверно замедляло темп нарастания нарушения внешнего дыхания у пациентов с БАС. Дозировка 3 мл и плацебо влияния на динамику не оказывало.
Необходимо отметить, что доза в 3 мл, которая была использована изначально и планировалась как стандартная, не показала каких-либо значимых изменений в состоянии больных. Но возможно, это было связано с тем, что при распределении участников исследования в эту группу попало очень много больных, которые имели, как оказалось впоследствии, достаточно агрессивные и быстро прогрессирующие темпы развития заболевания. Поэтому, возможно, это было связано не с самой дозой препарата, а со степенью нарастания неврологической симптоматики у некоторых пациентов.
Препарат АдеВаск не влиял на динамику показателей ночной пульсоксиметрии и на уровень десатурации. При оценке мышечной силы с использованием кистевого динанометра положительный эффект был достигнут только у группы пациентов-мужчин, которые получали препарат в дозе 1,5 мл. Поэтому говорить о том, что препарат оказывает влияние на изменение двигательных активностей и силы мышц кисти, некорректно.
Одной из переменных эффективности был перевод на неинвазивную вентиляцию легких или установка гастростомы. В течение всего периода наблюдений всего 2 человека были переведены на неинвазивную вентиляцию легких. Это были пациенты из группы, получавших АдеВаск в дозе 1,5 мл. На гастростомию в течение исследования попало 7 человек. Достоверной разницы по дозам препаратов, по сравнению с плацебо, при переводе на гастростому получено не было.

Летальный исход в течение нашего исследования наступил у трех пациентов из групп АдеВаск 1,5 мл, АдеВаск 6 мл и плацебо. В группе АдеВаск 3 мл, несмотря на наличие достаточно большого количества быстропрогрессирующих пациентов, летального исхода не было зафиксировано. Все группы статистически не различались по выживаемости пациентов, поэтому достоверно оценить выживаемость в нашем исследовании не представлялось возможным. Это было связано с небольшим количеством участников (всего 100 человек) и с короткой продолжительностью исследования. Например, в исследовании рилузола, которое показало увеличение выживаемости пациентов, участвовало около тысячи человек, а длилось оно 18 месяцев.
За время нашего исследования у 100 пациентов на 1200 введений препарата развилось 291 нежелательное явление (то есть на каждое четвертое введение). Все они были разделены на две большие группы — местные и системные.
Местные нежелательные явления описывались пациентами, в основном, как покраснения, отеки, боли, реакции в месте инъекции, уплотнение, зуд, сыпь. В одном случае возник периферический отек конечности. Стоит отметить, что в течение от одного до четырех дней мы отмечали полный регресс данных нежелательных явлений.
К системным явлениям относилась самая основная и ожидаемая реакция — это гриппоподобная реакция, которая пациентами описывалась в виде лихорадки, астении, чувства усталости, озноба, гипертермии, дискомфорта в груди. Степень их проявления была от умеренной до выраженной. Эти явления также носили временный характер. В одном случае мы отмечали генерализованный отек конечностей. Это возникло при использовании препарата в дозе 6 мл.
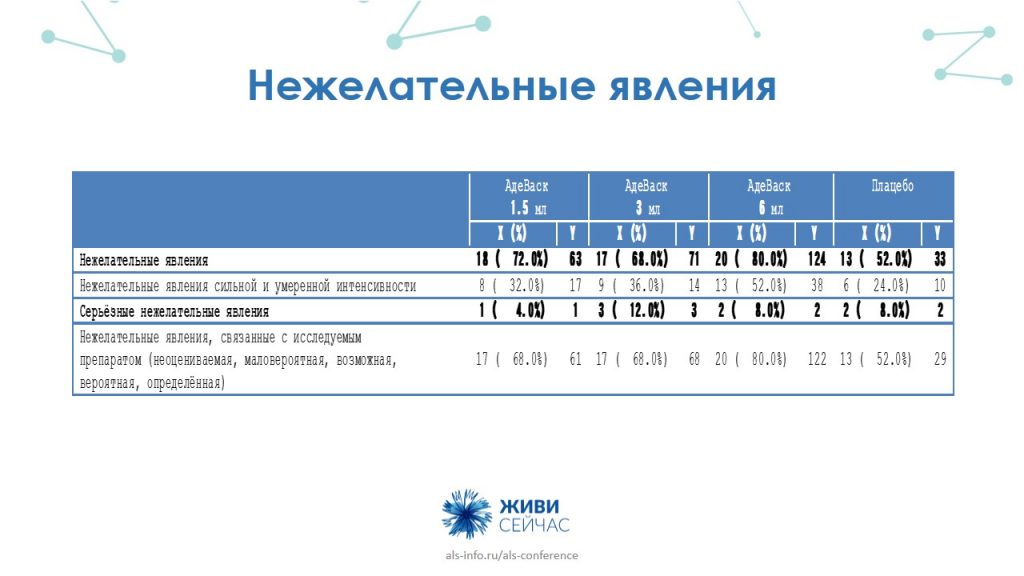
Проявившиеся нежелательные явления носили дозозависимый эффект. Наибольшее количество нежелательных явлений мы наблюдали у пациентов, которые получали препарат в дозе 6 мл. Но и эффективность была показана также у пациентов именно этой группы.
За все время исследования мы получили 8 серьезных нежелательных явлений. В группе 1,5 мл — это дыхательная недостаточность у одного пациента. В группе 3 мл было получено 3 нежелательных серьезных явления: почечная колика, сосудистый коллапс и выраженная гипертензия. В группе 6 мл — 2 серьезных нежелательных явления: кардиопульмональная и дыхательная недостаточность. В группе плацебо зафиксировали 2 серьезных явления. Стоит отметить, что бо́льшая часть этих серьезных нежелательных явлений, таких как дыхательная недостаточность, стала следствием естественного течения заболевания. Поэтому связи с исследуемым препаратом получено не было. Развитие почечной колики также не было достоверно связано с введением исследуемого препарата. В итоге по частоте и характеру серьезных нежелательных явлений группы АдеВаск не отличались от группы плацебо.
Выводы

На основании полученных данных мы смогли сделать выводы, что препарат вызывает характерные системные реакции в виде гриппоподобного синдрома и местные реакции в местах введения препарата. Большинство нежелательных явлений, которые мы наблюдали при применении препарата АдеВаск, были ожидаемыми. Они характеризовались преимущественно слабой или умеренной степенью тяжести. Их частота имела дозозависимый эффект: бо́льшая доза препарата вызывает более частые и более выраженные нежелательные явления.
Практически все наблюдавшиеся серьезные нежелательные явления не имели связи с исследуемым препаратом или были следствием естественного течения заболевания. Применение препарата АдеВаск во всех исследованных дозировках у пациентов с БАС было безопасным. Препарат продемонстрировал благоприятный профиль нежелательных явлений. Использование АдеВаск в дозе 6 мл замедляло темп прогрессии неврологического дефицита, который оценивался с помощью шкалы ALSFRSR. Применение АдеВаск в дозе 1,5 и 6 мл замедлял темп нарастания нарушения внешнего дыхания у пациентов с БАС при оценке отношения функции внешнего дыхания к должному объему легких.
Выводом является то, что препарат именно в дозе 6 мл показал свою безопасность и эффективность у пациентов с БАС. Но говорить о том, что препарат работает, можно с большими оговорками. Многие пациенты по разным причинам не завершили полностью участие в исследовании, которое подразумевало 17 введений. Это и нарастание неврологического дефицита, и невозможность приезжать в наш центр для повторного введения препарата. Поэтому были учтены не все пациенты, которые изначально включались в исследование.
На данный момент (по состоянию на 6 апреля 2019 года — Прим. ред.) проводится экспертиза качества препарата в Минздраве. В скором времени будут поданы документы на ускоренную регистрацию данного препарата, учитывая тяжесть и редкость заболевания, отсутствие препаратов для терапии данной патологии в России.
На сегодняшний момент площадки для производства данного препарата в промышленных масштабах не существует: компания-производитель ведет ее активный поиск. Поэтому указать конкретные сроки появления препарата на рынке достаточно сложно.
Вопросы из зала

Было ли исследование слепым, знали ли пациенты что им вводят — плацебо или препарат? Как вводились большие дозы — по 1,5 мл или больше?
Васильев А. В.: Да, исследование было заслеплено — пациенты не знали о том, что и в какой дозе они получают. Препарат вводился однократно: были использованы шесть точек введения — это мышцы бедра, дельтовидные и трапециевидная мышца. То есть в каждую мышцу, соответственно, вводилась одна шестая часть препарата. В группе АдеВаск 6 мл — по 1 мл в каждую точку вкола; в группе АдеВаск 1,5 мл — по 0,25 мл; в группе плацебо —по 0,5 мл.
Были ли среди участников исследования пациенты с бульбарной формой?
Васильев А. В.: Да, были как со спинальным дебютом, так и с бульбарными нарушениями. Но эффекта при бульбарных формах мы не получили.
Если это лекарство будет принято и выпущено, вы бы рекомендовали его принимать отдельно или вместе с рилузолом?
Васильев А. В.: Это сложный вопрос. В нашем исследовании не было показано влияние данного препарата на функцию печени, в первую очередь. У нас было несколько пациентов на более поздних стадиях, которые подключали к терапии рилузол. Они не были, конечно, учтены из-за протокола: он запрещает использование препаратов, которые могут как-то дополнительно влиять на оценку результатов. В будущем в рамках исследований можно попробовать их совместить, не вижу предпосылок для того, чтобы разделять их прием.
Благодарим за помощь в расшифровке Ануш Сулейманян.